重症肌无力的诊疗攻略及研究进展
重症肌无力(myastheniagravis,MG)是由自身抗体介导的获得性神经-肌肉接头(neuromuscularjunction,NMJ)传递障碍的自身免疫性疾病。
乙酰胆碱受体(AChR)抗体是MG最常见的致病性抗体;此外,针对突触后膜其他组分,包括:肌肉特异性受体酪氨酸激酶(MuSK)、低密度脂蛋白受体相关蛋白4(LRP4)及兰尼碱受体(RyR)等抗体陆续被发现参与MG发病,这些抗体可干扰AChR聚集、影响AChR功能及NMJ信号传递[1]。
本文将梳理重症肌无力的诊断方式和常见治疗方法。其诊断方式包括:临床表现、辅助检查、诊断标准、鉴别诊断;其治疗方法包括:急性加重期治疗、药物治疗、外科治疗。
诊断方式
其诊断依据包括:临床表现、辅助检查、诊断依据、鉴别诊断。
临床表现
全身骨骼肌均可受累的一种病症表现为波动性无力和易疲劳性,呈现“晨轻暮重”特征,活动后症状加重,休息后减轻。眼外肌最易受累,常为首发症状,表现为上睑下垂和/或双眼复视。面肌、咀嚼肌、咽喉肌、颈肌及肢体肌肉均可受累,导致多种症状如眼睑闭合无力、鼓腮漏气、构音障碍、吞咽困难、抬头困难及肢体活动困难等。病情常从一组肌群开始,逐渐累及其他肌群,直至全身肌无力。部分患者病情进展迅速,可能出现肌无力危象。
辅助检查
药理学检查
甲硫酸新斯的明试验用于评估肌无力,成人剂量1.0~1.5mg,儿童按体重0.02~0.04mg/kg(最大1.0mg),同时注射阿托品0.5mg以减少M胆碱样不良反应。试验前,根据MG临床绝对评分标准,选取症状最明显肌群记录肌力。注射后,每10分钟记录一次,持续60分钟。以改善最显著时的单项绝对分数,通过下列公式计算相对评分:相对评分=(试验前评分-注射后每次评分)/试验前评分×100%。结果判定:小于等于25%为阴性,25%~60%为可疑阳性,大于等于60%为阳性。
电生理检查
重症肌无力的常用电生理检查包括:重复神经电刺激、单纤维肌电图。
重复神经电刺激
重复神经电刺激(repetitivenervestimulation,RNS)采用低频电刺激神经干,记录相应肌肉的复合肌肉动作电位(compoundmuscleactionpotential,CMAP),常规检测面神经、副神经、腋神经和尺神经。如果第4或第5波与第1波波幅比值衰减超过10%,称为波幅递减,部分患者呈现U字样改变。服用胆碱酯酶抑制剂者需停药12~18小时后检查,但需权衡病情。为鉴别突触前膜病变,可进行高频RNS或大力收缩后观察CMAP波幅变化,递增100%以上为异常,称为波幅递增。
单纤维肌电图
单纤维肌电图(singlefiberelectromyogram,SFEMG)使用特殊针电极测量同一神经肌纤维电位间隔,反映神经肌肉接头的功能。如果“颤抖”(Jitter)超过55μs为异常,记录20个中有2个及以上大于55μs则判定为异常,阻滞(block)也判定为异常。SFEMG的敏感性高,不受胆碱酯酶抑制剂影响,主要用于眼肌型重症肌无力(ocularmyastheniagravis,OMG)或临床怀疑MG但RNS未见异常的患者。
血清抗体检测
MG的相关抗体检测包括:抗AChR抗体、抗MuSK抗体、抗LRP4抗体、抗横纹肌抗体。
抗AChR抗体:约50%~60%的OMG、85%~90%的GMG血清中可检测到,放射免疫沉淀法(RIA)为标准方法,ELISA法的敏感性较低。阴性结果不能排除MG诊断。
抗MuSK抗体:在10%~20%的AChR抗体阴性MG患者血清中可检测到,RIA或酶联免疫吸附试验(ELISA)为标准方法。
抗LRP4抗体:7%~33%的AChR、MuSK抗体阴性MG患者可检测出,用于进一步诊断。
抗横纹肌抗体:包括抗Titin和RyR抗体,前者常用ELISA法检测,后者可采用免疫印迹法或ELISA法检测,用于MG的辅助诊断。
胸腺影像学检查
大约80%左右的MG患者伴有胸腺异常,包括胸腺增生及胸腺瘤。CT为常规检测胸腺方法,胸腺瘤检出率可达94%;MRI有助于区分一些微小胸腺瘤和以软组织包块为表现的胸腺增生;必要时可行CT增强扫描;PET-CT有助于区别胸腺癌和胸腺瘤。
合并其他自身免疫性疾病检测
MG患者可合并其他自身免疫疾病,如自身免疫性甲状腺疾病,最常见的是Graves病,其次为桥本甲状腺炎[2]。OMG合并自身免疫性甲状腺疾病比例更高,因此,MG患者需常规筛查甲状腺功能及甲状腺自身抗体、甲状腺超声检查观察有无弥漫性甲状腺肿大,以及其他自身免疫性疾病相关抗体检测。
诊断依据
在具有典型MG临床特征(波动性肌无力)的基础上,满足以下3点中的任意一点即可做出诊断,包括:药理学检查、电生理学特征以及血清抗AChR等抗体检测。同时需排除其他疾病。所有确诊MG患者需进一步完善胸腺影像学检查(纵隔CT或MRI),进一步进行亚组分类。
鉴别诊断
与眼肌型重症肌无力(ocularmyastheniagravis,OMG)的鉴别诊断(详见表1)[3]:
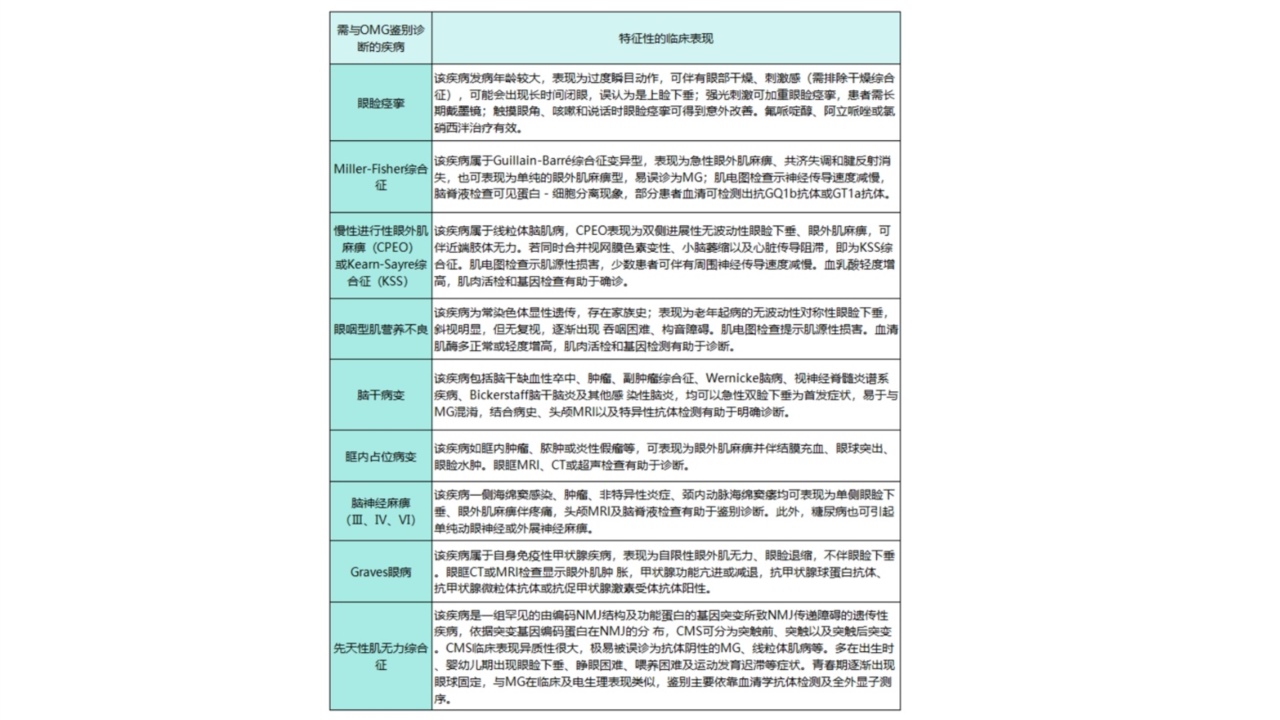
表1:需要与OMG鉴别诊断的疾病
与全身型重症肌无力(generalizedmyastheniagravis,GMG)的鉴别诊断(详见表2):
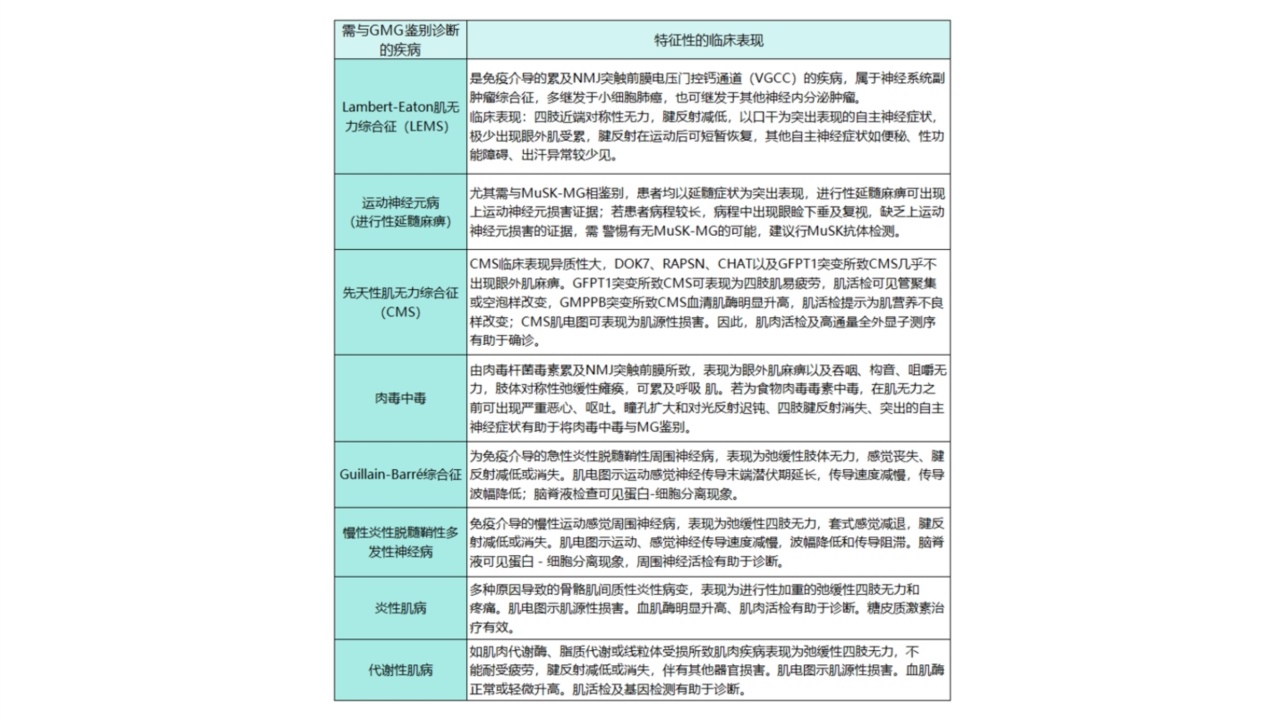
表2:需要与GMG鉴别诊断的疾病
常见的治疗方法
目前,MG的治疗仍然以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白(IVIG)、血浆置换(PE)以及胸腺切除为主。
急性加重期治疗
IVIG与PE主要用于病情快速进展、危及生命的情况,如肌无力危象、严重的球麻痹所致吞咽困难、肌无力患者胸腺切除术前和围手术期治疗,可使绝大部分患者的病情得到快速缓解。为达到持续缓解,可同时启动免疫抑制治疗(非激素类免疫抑制剂),由于激素早期可一过性加重病情,甚至诱发肌无力危象,于IVIG与PE使用后症状稳定时添加激素治疗。IVIG多于使用后5~10天左右起效,作用可持续2个月左右。在稳定的中、重度MG患者中重复使用并不能增加疗效或减少糖皮质激素的用量。
药物治疗
胆碱酯酶抑制剂—症状性治疗
溴吡斯的明是治疗MG的一线药物,可缓解、改善大部分MG患者的临床症状,应作为初始治疗的首选,可与激素及其他免疫抑制剂联合使用。成人首次剂量60mg,3~4次/天,最大剂量480mg/天。其副作用包括:恶心、流涎等,妊娠期使用该药物是安全有效的。
免疫抑制治疗
免疫抑制药物包括糖皮质激素和其他口服非激素类免疫抑制剂,如硫唑嘌呤(AZA)、他克莫司(FK-506)、吗替麦考酚酯(MMF)、环孢素、甲氨蝶呤(methotrexate)及环磷酰胺(cyclophosphamide)。非激素类免疫抑制剂在糖皮质激素减量以及预防MG复发中发挥重要作用。但目前尚无临床研究比较不同非激素类免疫抑制剂的疗效,因此,药物的选择尚无统一标准,更多依赖于临床医生的经验。
靶向生物制剂
目前临床上用于MG治疗的靶向生物制剂包括已经被美国食品和药物监督管理局(FDA)批准使用的靶向补体的依库珠单抗(eculizumab),以及适应证外用药的靶向B细胞的利妥昔单抗(rituximab,RTX)。此外,一些靶向免疫系统不同组分的生物制剂仍在临床前研究,如靶向B细胞激活因子(BLyS)的Belimumab以及靶向FcRn的Ef-gartigimod等。
外科治疗
外科治疗作为MG全程管理的重要组成部分,需综合考虑患者的发病年龄、疾病严重程度、血清学特点、是否合并胸腺肿瘤、非手术治疗疗效、手术治疗指征、治疗并发症以及治疗费用等,做好围手术期处理,尽量做到安全、有效、精准化、个体化、微创化的治疗[4]。
根据最新美国神经病学会、欧洲重症肌无力诊治指南以及中国重症肌无力诊断和治疗专家共识,认为胸腺切除术是治疗MG的有效方法,早期手术对改善预后有积极作用和重要意义[5-6]。
但由于MG分型的多样性、胸腺区解剖位置的特殊性、手术方式多样性以及手术入路的可选择性等,胸腺切除术的适应证、手术时机、手术方式、手术径路、围手术期管理以及疗效评价等仍存在争议。
胸腺是MG患者产生致病抗体的器官,在诱导和维持MG患者AchR-Ab等抗体产生中起着关键作用,临床发现约10%~15%的MG患者存在胸腺瘤,约30%的胸腺瘤患者合并MG,约70%的MG患者存在胸腺增生等改变,因此,胸腺切除是治疗MG的有效手段之一[7]。胸腺的迁移方式复杂,异位胸腺广泛分布,可位于前后纵隔、颈部甚至腹部,手术需行相对的胸腺扩大切除术,减少异位胸腺残留的可能[8]。
研究进展
针灸治疗重症肌无力的系统评价和荟萃分析

重症肌无力(MG)是一种常见的自身免疫性疾病,通常涉及全身骨骼肌,严重影响患者的生活质量。针灸治疗MG具有得天独厚的优势,旨在评价针灸对MG的临床疗效。
该研究共纳入11项研究,涉及658例患者,其中治疗组330例,对照组328例。Meta分析结果显示,治疗组在提高总临床缓解率方面优于对照组。此外,治疗组在提高绝对临床评分方面优于对照组。治疗组与对照组在改善血清白细胞介素-6受体水平和OMG定量评分方面差异无统计学意义。对总临床有效率进行发表偏倚检验,结果显示漏斗图的两侧不对称,提示可能存在发表偏倚。针灸对MG的疗效较好,在提高临床总有效率和临床绝对评分方面优于常规西医[9]。
参考文献:
[1]中国免疫学会神经免疫分会,常婷,李柱一,etal.中国重症肌无力诊断和治疗指南(2020版)[J].中国神经免疫学和神经病学杂志,2021,28(1):12.DOI:10.3969/j.issn.1006-2963.2021.01.001.
[2]AminS,AungM,GandhiFR,PenaEscobarJA,GulraizA,MalikBH.MyastheniaGravisanditsAssociationWithThyroidDiseases.Cureus.2020Sep4;12(9):e10248.doi:10.7759/cureus.10248.PMID:33042687;PMCID:PMC7536109.
[3]EngstromJW.Myastheniagravis:diagnosticmimics.SeminNeurol.2004Jun;24(2):141-7.doi:10.1055/s-2004-830903.PMID:15257510.
[4]谭群友,陶绍霖,刘宝东,柳阳春.重症肌无力外科治疗中国临床专家共识[J].中国胸心血管外科临床杂志,2022,29(5):529-541
[5]中国免疫学会神经免疫分会.中国重症肌无力诊断和治疗指南(2020版).中国神经免疫学和神经病学杂志2021,28(1):1-12.
[6]MelzerN,RuckT,FuhrP,etal.Clinicalfeatures,pathogenesis,andtreatmentofmyastheniagravis:AsupplementtotheguidelinesoftheGermanNeurologicalSociety.JNeurol,2016,263(8):1473-1494.
[7]NarayanaswamiP,SandersDB,WolfeG,etal.Internationalconsensusguidanceformanagementofmyastheniagravis:2020update.Neurology,2021,96(3):114-122.
[8]MarxA,PfisterF,SchalkeB,etal.Thedifferentrolesofthethymusinthepathogenesisofthevariousmyastheniagravissubtypes.AutoimmunRev,2013,12(9):875-884.
[9]LuY,LiJ,YuT,WuC,YouY,WangC,LiuX.Acupunctureandmoxibustiontreatmentformyastheniagravis:Asystematicreviewandmeta-analysis.Medicine(Baltimore).2024May3;103(18):e37961.doi:10.1097/MD.0000000000037961.PMID:38701271;PMCID:PMC11062737.
编辑|麦麦
排版|麦麦
审核|梓霖
[声明:本网站所有内容,凡未注明来源为“转载”,版权均归巢内网所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:巢内网”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们]


相关推荐
